Covaris
A Robust and Standardized Workflow to Analyze C. albicans
Differential Protein Expression using Tandem Mass Tag (TMT)
Case Study
A Robust and Standardized Workflow to Analyze C. albicans
Differential Protein Expression using Tandem Mass Tag (TMT)

Covaris manufactures advanced sample preparation tools based on a unique technology platform and develop new pre-analytical sample preparation technologies to help accelerate the pace of research and drug discovery. Applications include faster automated DNA fragmentation, cell lysis, mixing, bead mixing and resuspension, and compound formulation.
As the analytical methods of proteomic analysis have become more sensitive, it has opened the field for the utilization of limited and difficult sample types. Thus, fungi samples are difficult to process for protein extraction.
They are resistant to chemical lysis and exhibit phenotypic appearances, which make them even harder to lyse, especially when it comes to the filamentous subtypes. Mechanical treatments can circumvent the use of multiple heating and cooling steps, but they must ensure an efficient and reproducible lysis.
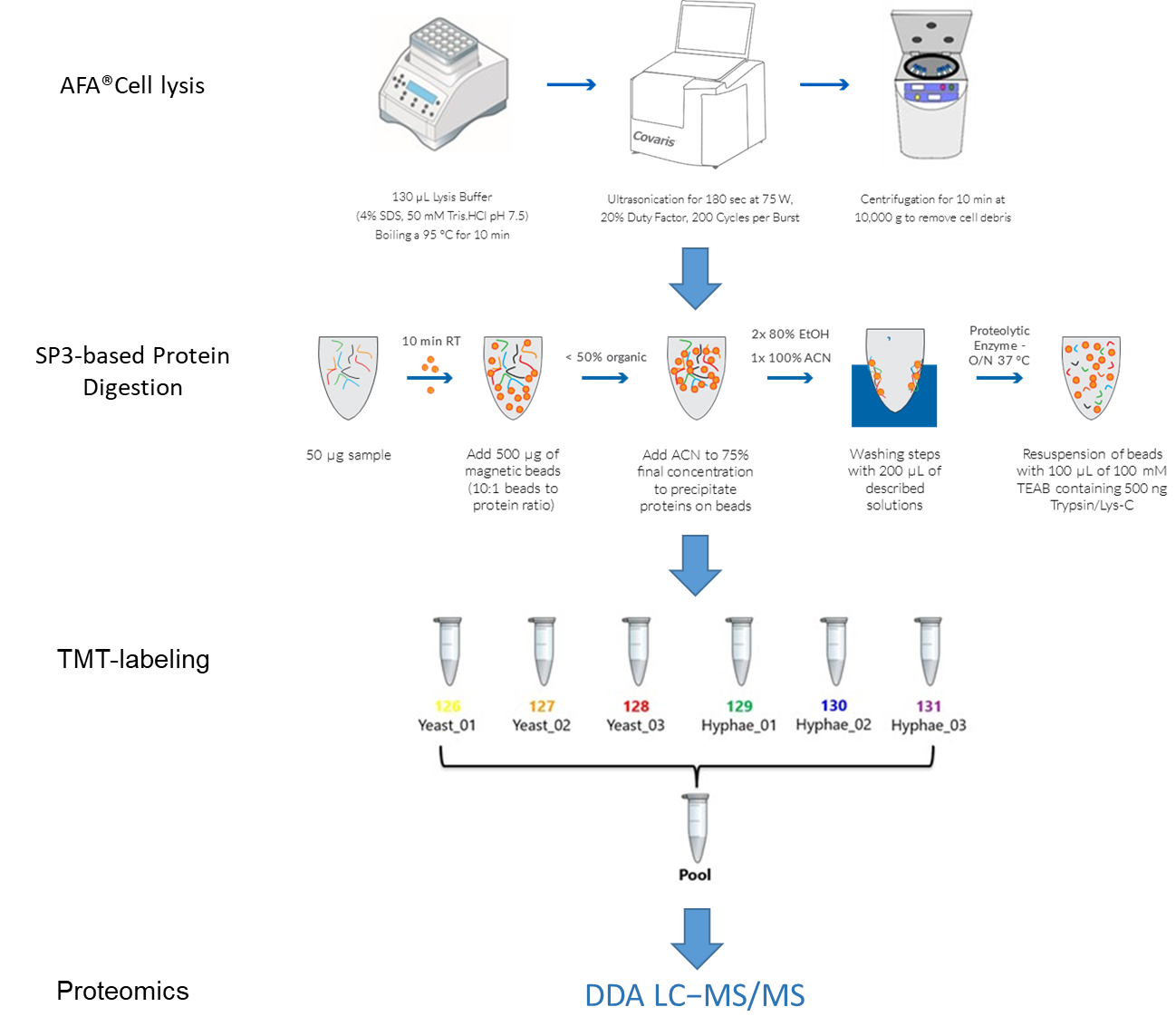
Based on sample preparation technologies developed by Covaris, Bioaster implemented a robust method for reproducible extraction and analysis of proteins from C. albicans, in both its yeast and hyphal forms. The process combines Adaptive Focused Acoustics® (AFA®) for the lysis, Single Pot Solid Phase Sample
Using the workflow implemented, 2933 proteins in total were identified by DDA LC-HRMS analysis, corresponding to 29,148 unique peptides.
A Principal Component Analysis (PCA), run on 6 Yeast and Hyphae samples, showed as expected a separation between the two phenotype, with the 3 Yeast samples clustered together, as well as the 3 Hyphae samples.
A volcano plot analysis of the data confirmed the differential expression and the protein preservation during the extraction. Thus, 65 proteins have been identified as upregulated and 130 as downregulated in the Hyphae type, in line with previous observations.
Thanks to this analytical workflow, we may address proteomics challenges with other kind of difficult samples. Moreover, this protocol can be easily adapted for automated, higher-throughput processing.